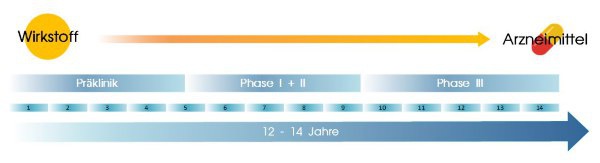
Von ursprünglich 10.000 untersuchten Wirkstoffen landet nach 12-14 Jahren Entwicklungs- und Registrierungszeit nur einer als Arzneispezialität auf dem Markt.
„Guten Tag, ich würde gerne dieses Rezept hier einlösen. Und außerdem hätte ich noch gerne eine Packung dieser brandneuen Tabletten gegen Reisekrankheit, die Sie mir letztes Mal empfohlen haben. Die Wirkung war ja phänomenal!“ – „Sehr gerne, das freut mich zu hören. Darf ich sonst noch etwas für Sie tun?“
Eine Szene, wie sie tagtäglich in jeder Apotheke zum Alltag gehört. Eine Fülle an Medikamenten, fein säuberlich sortiert in Regalen, Laden und Auszügen, steht bereit, Patienten zu helfen, Leiden zu lindern, Krankheiten zu heilen. Doch was vielen oft gar nicht bewusst ist: Bis hierher war es für jedes einzelne von ihnen ein langer Weg. Viel Zeit, Know-how und Ressourcen wurden investiert. Aber wie kommt die Tablette eigentlich in den Apothekerschrank?
Ein langer Weg
Die Geschichte eines neuen Medikaments beginnt vor ungefähr 10 bis 14 Jahren in einem Labor. Auf der Suche nach neuen Wirkstoffen durchforsten Wissenschaftler Datenbanken und Fachliteratur. Mit den aussichtsreichsten Substanzen werden erste Tests durchgeführt. Nur bei ca. 12 von 5.000 bis 10.000 Wirkstoffen lohnt es sich, weitere Forschungen anzustellen. Sie erreichen die ‚präklinische Phase‘.
Nun werden die Wirkstoffe an menschlichen Zellen getestet um herauszufinden, ob sich die erhoffte Wirkung auch tatsächlich einstellt. Gleichzeitig werden Tierversuche mit Mäusen, Ratten oder Kaninchen durchgeführt um die Wirkung der eingesetzten Substanzen auf deren Stoffwechsel zu untersuchen. Etwa 21% der Forschungs- und Entwicklungskosten eines neuen Arzneimittels entfallen auf diese Phase. Ungefähr drei von zwölf Wirkstoffen schaffen die Hürde der präklinischen Studien nicht, da sie sich als unwirksam oder möglicherweise sogar schädlich erweisen.
Die restlichen neun Wirkstoffe werden in den darauffolgenden klinischen Studien dann erstmals am Menschen getestet. Auf die drei einzelnen Phasen dieses Forschungsstadiums entfällt gut die Hälfte der gesamten Arzneimittel-Entwicklungskosten. In Phase I werden die Substanzen 10 bis 20 gesunden Freiwilligen verabreicht. Anhand von Blut- und Urinproben der Probanden lässt sich feststellen, wie die Wirkstoffe vom menschlichen Organismus aufgenommen, verarbeitet und ausgeschieden werden.
„Survival of the Best“
Nur etwa fünf von neun Wirkstoffen erreichen Phase II der klinischen Studien. Nun werden die Substanzen bei kranken Menschen eingesetzt um herauszufinden, ob die erhoffte Wirkung bei den Patienten auch tatsächlich eintritt. Gleichzeitig werden eventuelle Nebenwirkungen dokumentiert und in mehreren Testreihen die optimalen Dosierungen für die Therapie ermittelt. Drei von fünf Wirkstoffen scheitern während dieser Testphase.
Die restlichen Wirkstoffe werden schließlich in Phase III weltweit an mehreren tausend Patienten erprobt. Dabei wird ihre therapeutische Wirksamkeit unter anderem in placebokontrollierten Studien unter Beweis gestellt. Ist auch diese Hürde gemeistert – im Schnitt bleibt hier schlussendlich nur mehr ein einziger Wirkstoff übrig - steht einer Einreichung zur Zulassung nichts mehr im Wege.
Der letzte Schritt: die Zulassung
Als Zulassungsverfahren kommen entweder das "Nationale Verfahren", das "Dezentrale Verfahren" bzw. "Verfahren der gegenseitigen Anerkennung" oder das "Zentrale Verfahren" in Frage. Während bei der Nationalen Zulassung das Arzneimittel nur im Bereich des zulassenden Staates in Verkehr gebracht werden darf, wird bei der Zentralen Zulassung durch die EU-Kommission die Zulassung für sämtliche Staaten der EU ausgesprochen. Liegt bereits eine Erstzulassung in einem EU-Land vor, so können andere EU-Länder auf dieser Grundlage das Arzneimittel in einem etwas vereinfachten Verfahren zulassen (Dezentrale Zulassung).
Nun muss der Antragsteller des neuen Arzneimittels durch Vorlage pharmazeutischer, präklinischer und klinischer Daten im sogenannten Zulassungsdossier nachweisen, dass der zu erwartende Nutzen des Medikaments die unerwünschten Nebenwirkungen übersteigt und der entdeckte Stoff auch tatsächlich wirkt. Außerdem wird im Zulassungsverfahren unter anderem der Text der Fachinformation, der Text der Gebrauchsinformation, die Beschriftung der Verpackung, der Rezeptpflichtstatus und der einzuhaltende Distributionsweg überprüft und festgelegt. Sämtliche Prüfungsergebnisse werden in schriftlichen Gutachten dokumentiert. Wurden alle Gutachten positiv bewertet, wird der Zulassungsbescheid ausgestellt und das neue Medikament ist bereit für den sogenannten „Launch“.
Von ursprünglich 10.000 untersuchten Wirkstoffen landet nach 14 Jahren Entwicklungs- und Registrierungszeit nur einer als Arzneispezialität auf dem Markt – und in Ihrem Apothekerschrank.
Quellen: