Bei der Moyamoya-Erkrankung bildet sich ein wolkenartiges Kollateralnetzwerk in Hirngefäßen aus. Die ungewöhnliche Krankheit kann Schlaganfälle auslösen und betrifft in Deutschland nur wenige Menschen – doch die Dunkelziffer ist hoch.
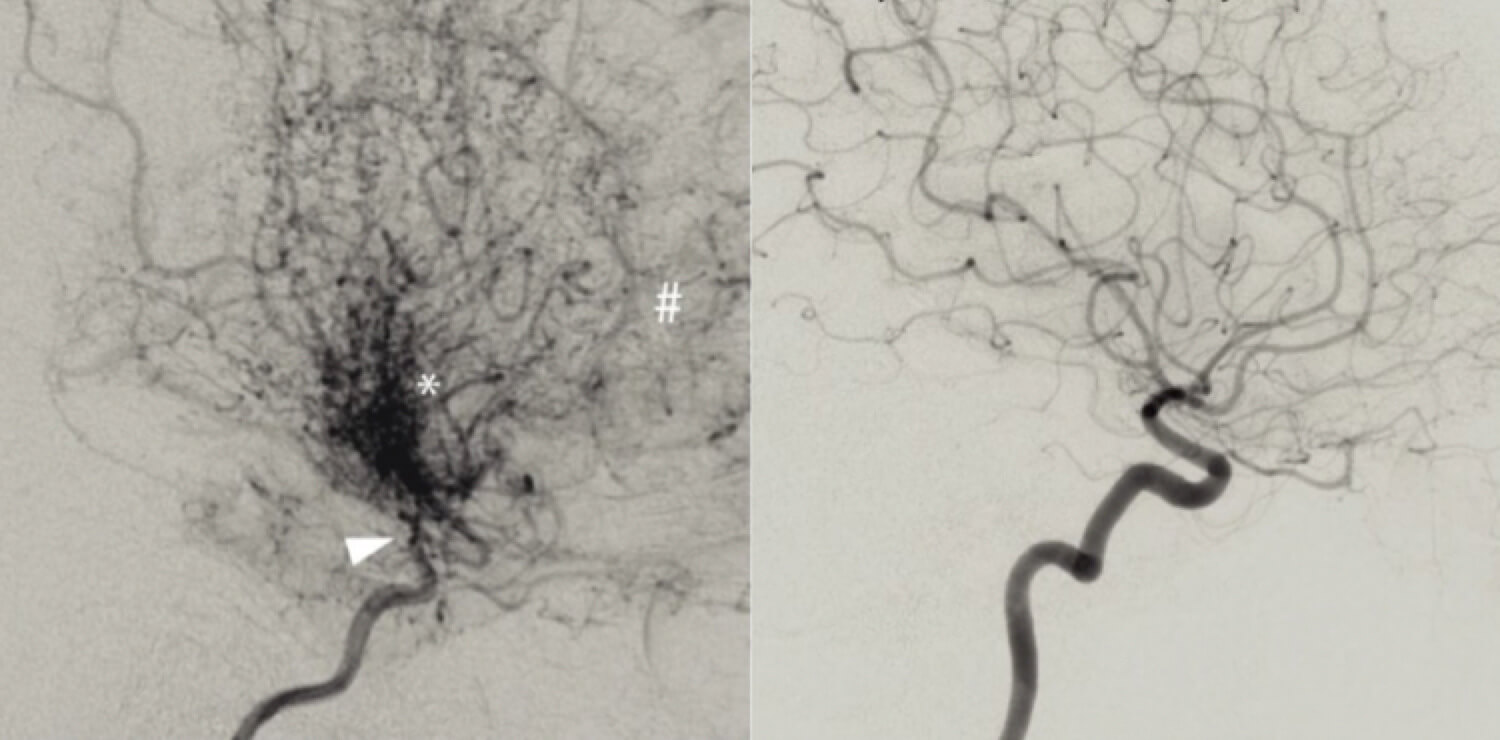
Im Rahmen der Camp Woche zu Orphan Diseases wollen wir den Fokus heute auf eine ungewöhnliche Schlaganfallursache legen – die Moyamoya-Erkrankung. Hierbei handelt es sich um eine sehr seltene Gefäßerkrankung, die zu beidseitigen progredienten Verengungen der terminalen A. carotis interna und ihren Aufzweigungen im Circulus Willisii (A. cerebri media und anterior) führt. Hierbei bildet sich das für die Erkrankung typische wolkenartige Kollateralnetzwerk aus, welches ihr den Namen Moyamoya (zu Deutsch etwa: „Wölkchen“ oder „Nebel“) einbrachte.
Die pathophysiologischen Mechanismen hinter diesem Prozess sind bis heute nicht gänzlich geklärt. Bislang weiß man, dass es zu einer Verdickung der Gefäßintima und so zur zunehmenden Gefäßstenosierung und Ausbildung von fragilen Kollateralgefäßen kommt.
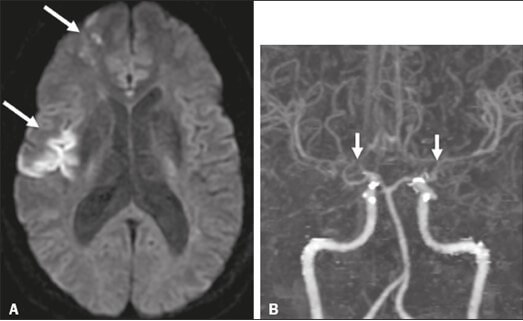
Die Erkrankung tritt vor allem im asiatischen Raum auf und ist in Europa deutlich seltener (ca. 0,03/100.000), es wird jedoch eine hohe Dunkelziffer vermutet, da die Diagnosestellung aufgrund der Seltenheit erschwert wird. In etwa 60 % der Fälle wird initial eine falsche Diagnose gestellt. Typischerweise manifestiert sich die Erkrankung erstmals im Kindes- oder jungen Erwachsenenalter. Symptome können passagere neurologische Defizite im Sinne von TIAs sein, aber auch juvenile Schlaganfälle mit schweren bleibenden neurologischen Ausfällen sind möglich. Zudem werden Kopfschmerzen und zunehmende kognitive Defizite beschrieben.
Meist erfolgt bei den Patienten zunächst ein cMRT mit MR-Angiografie, welches in der Regel zahlreiche ischämische Defekte bei gleichzeitigem Vorliegen von Hauptstammstenosen der Hirngefäße mit Kollateralnetzwerk zeigt. Bei Verdacht auf eine Moyamoya-Erkrankung sollte unbedingt eine digitale Subtraktionsangiographie (DSA) ergänzt werden, da so das typische wolkenartige Kollateralnetzwerk zur Darstellung kommt. Auch die transkranielle Dopplersonographie sowie Perfusionsmessungen (MR-Perfusion, PET, SPECT) können bei der Diagnosestellung hilfreich sein. Eine Lumbalpunktion wird empfohlen, um eine potenziell therapierbare, entzündliche Erkrankung auszuschließen.
Bei initialer Diagnosestellung beginnt man häufig, je nach Blutungsgefahr, zunächst eine Sekundärprophylaxe mit ASS 100 mg. Zudem sollte auf eine gute Blutdruckeinstellung geachtet werden, da dieser weder zu hoch noch zu niedrig sein darf. Bei hohem Druck kann es zu Schädigung der Kollateralen und hierdurch zu Hirnblutungen kommen. Ein niedriger Blutdruck wiederum verringert die Hirndurchblutung und kann somit zu ischämischen Schlaganfällen führen.
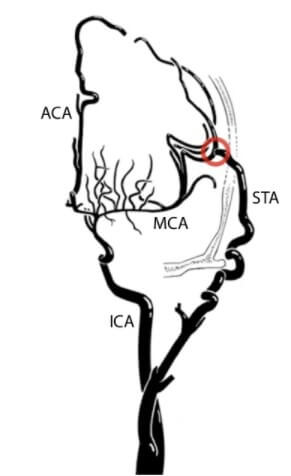
Bei schnell fortschreitenden Gefäßveränderungen oder wiederholten ischämischen Ereignissen besteht die Indikation zur operativen Therapie. Meist wird hierbei ein direkter Bypass zwischen der A. temporalis superficialis und der stenosierten A. cerebri media hergestellt. Dieses Verfahren erfordert eine hohe Expertise des durchführenden Chirurgen und sollte nur in spezialisierten Zentren durchgeführt werden. Bei Kindern sind die Gefäße durch ihre kleine Größe für diese Art der Operation oft ungeeignet. Man behilft sich dann mit einem indirekten Bypass. Hierbei wird etwa der gut vaskularisierte Temporalmuskel direkt auf das Hirn aufgelegt (Enzephalomyosynangiosis, EMS). Durch dieses Verfahren sollen Muskelgefäße ins Hirngewebe einsprießen, um dieses mit Blut zu versorgen.
Eine kausale Therapie existiert bislang noch nicht. Im Laufe der letzten Jahre gelang es den Forschern jedoch mehr und mehr Gene zu identifizieren, die an der Pathogenese der Erkrankung beteiligt sind. Hierzu gehört allen voran RNF213, welches insbesondere in der asiatischen Bevölkerung verbreitet ist und auch nur bei dieser zur Ausbildung einer Moyamoya-Erkrankung führen kann. Bei Kaukasiern konnte dies bislang nicht belegt werden. Es konnten jedoch in den letzten Jahren zahlreiche weitere Genvarianten identifiziert werden, welche auch bei europäischen Moyamoya-Patienten vorkommen.
Zudem deuten neue Forschungsergebnisse auf eine Assoziation zum HLA-System hin, einer Gengruppe, die für wesentliche Funktionen des menschlichen Immunsystems verantwortlich ist. Da viele dieser Genvarianten auch bei gesunden Individuen vorkommen, geht man von einem zusätzlichen Einfluss von Umweltfaktoren aus, welche bislang nicht identifiziert werden konnten.
Diese Faktoren zu identifizieren sowie die Erforschung neuer und bestehender Genvarianten bahnen den Weg für neue Therapiemöglichkeiten. Die Geschwindigkeit, in der in den vergangenen Jahren immer neue Genvarianten entdeckt und deren Beteiligung am Pathomechanismus identifiziert werden konnte, weckt Hoffnung, dass in naher Zukunft auch für die Moyamoya-Erkrankung eine kausale Therapie zur Verfügung steht.
Quellen
DOI: 10.1007/s12975-021-00940-2
Müllers, M. Zoom: Moyamoya-Angiopathie. DGNeurologie 5 (02/2022).
DOI: 10.1590/0100-3984.2021.0010
DOI: 10.1186/s13023-022-02238-4
https://doi.org/10.3892/etm.2019.7198
https://doi.org/10.3389/fneur.2021.687088
DOI: 10.1080/09084282.2012.721147
Bildquelle: Jelleke Vanooteghem, Unsplash