Eine beidseitige Parotis-Schwellung, die nicht wehtut, gibt den Ärzten ein Rätsel auf. Ist es Krebs oder doch was ganz anderes? Bei der DGIM-Tagung berichtet einer der behandelnden Ärzte von dem Fall.
Bei der DGIM 2022 Tagung in Wiesbaden berichtete Dr. Torsten Witte von der Rheumatologie der Medizinischen Hochschule Hannover von einem erinnerungswürdigen Patientenfall. Bei der Patientin handelte es sich um eine 52-jährige Frau, der seit rund einem Jahr eine langsam zunehmende Schwellung bei den Ohrspeicheldrüsen aufgefallen war. Die Schwellung war schmerzfrei, die Patientin hatte auch kein Fieber.
In der Vorgeschichte ließ sich ein unklarer Hautausschlag eruieren, der seit zwei Jahren bestand, nicht juckte und der ebenfalls allmählich zugenommen hatte. Zwei Jahre zuvor war zudem ein Exophthalmus ophthalmologisch abgeklärt worden, der danach rückläufig, zum aktuellen Vorstellungszeitpunkt aber noch latent vorhanden war. Die Ophthalmologen hatten per MRT eine linksseitige Dakyroadenitis diagnostiziert. Anamnestisch berichtete die Patientin ferner über eine „chronische Mastitis mit Zysten“ sowie ein seit drei Jahrzehnten bestehendes Asthma mit Rhinokonjunktivitis.
Von ambulant an die MHH-Ambulanz zugewiesen wurde die Patientin zur Abklärung der Verdachtsdiagnose eines Sjögren-Syndroms. Entsprechend wurde primär Blut entnommen, das nicht nur in der Standardpalette, sondern auch bei den rheumatologischen Parametern geradezu vorbildlich unauffällig war. Außer einer IgE-Erhöhung auf 453 IE/ml (Norm < 100) fand sich nichts. ANCA und ANA waren genauso wenig erhöht, wie die unter dem Verdacht auf Morbus Sjögren abgenommenen SSA/Ro- und SSB/La-Antikörper. Rheumafaktor, CCP und andere Entzündungsparameter? Normal.
Guter Rat war da erstmal teuer. „Die beste Idee wäre wahrscheinlich eine Parotis-Biopsie gewesen. Wir haben die Haut genommen, weil wir die Hautstanzen selbst vornehmen können“, so Witte. Die Stanze war dann zunächst auch nicht zielführend: Der Pathologe diagnostizierte unspezifische Veränderungen ohne gesteigerte proliferative Aktivität und äußerte den Verdacht auf ein autoimmunologisches Geschehen. Und jetzt?
Als erfahrene Rheumatologen hatten Witte und Kollegen glücklicherweise einen Verdacht, der sich letztlich als goldrichtig herausstellte. Da eine primär maligne Erkrankung aufgrund des symmetrischen Geschehens und des pathologischen Befunds als unwahrscheinlich angesehen werden konnte, wurde der Pathologe um einige immunologische Spezialfärbungen gebeten – insbesondere, unter dem Verdacht auf eine IgG4-assoziierte Erkrankung, um eine IgG4-Spezialfärbung. Dabei zeigten sich dann Infiltrate IgG-exprimierende Plasmazellen, die zu mehr als 50 Prozent IgG4-positiv waren. Auch im Serum war das IgG4 mit 3,89 g/l gegenüber einem Normwert von < 1,4 g/l deutlich erhöht. Entsprechend lautete die Abschlussdiagnose IgG4-assoziierte Erkrankung.
Die IgG4-assoziierte Erkrankung hat nicht nur keinen vernünftigen Namen, man weiß auch relativ wenig über sie. Die meiste Forschung dazu kommt aus Japan, wo Epidemiologen die Prävalenz auf 1 pro 18.000 schätzen, mit einem Erstmanifestationsalter von typischerweise jenseits des 50. Lebensjahrs. Männer gelten mit einem Verhältnis von 3 zu 1 als häufiger betroffen als Frauen. „Wahrscheinlich ist die Erkrankung aber viel häufiger“, so Witte.
Das klinische Bild ist extrem vielschichtig, und entsprechend schwierig kann die Erkrankung zu diagnostizieren sein.
Die Tränendrüsenschwellungen hören auf den Eigennamen Mikulicz-Syndrom, die Speicheldrüsenschwellungen hießen mal Küttner-Tumore. Ist die Schilddrüse befallen, sprechen manche von Riedel-Thyreoiditis. Sehr typisch ist eine autoimmune Pankreatitis Typ 1, und immerhin 25 % bis 50 % der Patienten haben die meist in der Urogynäkologie verortete, retroperitoneale Fibrose namens Morbus Ormond. Lunge, Prostata, Nieren, Gallenwege und Hypophyse können ebenfalls betroffen sein.
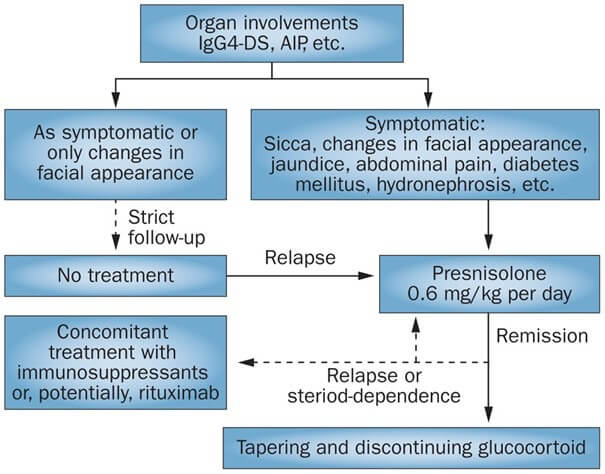
Therapeutisch steht bei der IgG4-assoziierten Erkrankung, je nach individuellem Leidensdruck beziehungsweise Ausgeprägtheit des individuellen Befunds, die ganze Palette von Abwarten bis „immunologisch voll draufhauen“ zur Verfügung. In einem japanischen Review wird ein Behandlungsalgorithmus vorgeschlagen:
Initial geben die Japaner Prednisolon 0,6 mg/kg, das dann nach Remission langsam versucht wird, auszuschleichen. Eine Studie aus dem Jahr 2019 zeigte, dass die initiale Kombination aus Glukokortikoid und Mycophenolat Mofetil Vorteile hat: Nach einem Monat war die Effektivität zwar gleich, aber MMF halbierte die Zahl der Rückfälle. Spätestens bei refraktärer Erkrankung kommt in der Regel die B-Zell-Depletion mit Rituximab zum Einsatz, die sehr effektiv ist, aber ebenfalls mit Rückfällen einhergehen kann, die dann in der Regel auf erneutes Rituximab wieder ansprechen. Gezeigt wurde das unter anderem in einer europäischen Studie beim Pankreatitis-Phänotyp.
Bildquelle: Ante Hamersmit, Unsplash