Bei der Mehrzahl der malignen Darmtumoren handelt es sich um Adenokarzinome, die aus den Drüsenzellen der Darmschleimhaut im Kolon oder Rektum entstehen.1,2 Da Kolon- und Rektumkarzinome viele Gemeinsamkeiten in Bezug auf ihre Ätiologie und Histologie haben, werden sie auch unter dem Begriff kolorektale Karzinome (CRC) zusammengefasst. Allerdings gibt es Unterschiede bei den Behandlungsstrategien.3,4
Im Jahr 2022 erkrankten weltweit mehr als 1,9 Millionen Menschen neu an einem kolorektalen Karzinom. Damit ist das CRC die dritthäufigste Krebserkrankung der Welt und steht an Platz zwei der Krebs-Todesursachen.5 In Deutschland betrifft etwa jede neunte Krebserkrankung das Kolon bzw. das Rektum.6 Das Risiko, an einem kolorektalen Karzinom zu erkranken, steigt mit dem Lebensalter an. So erkranken mehr als die Hälfte der Patient:innen jenseits des 70. Lebensjahres, wobei Männer häufiger betroffen sind als Frauen.6
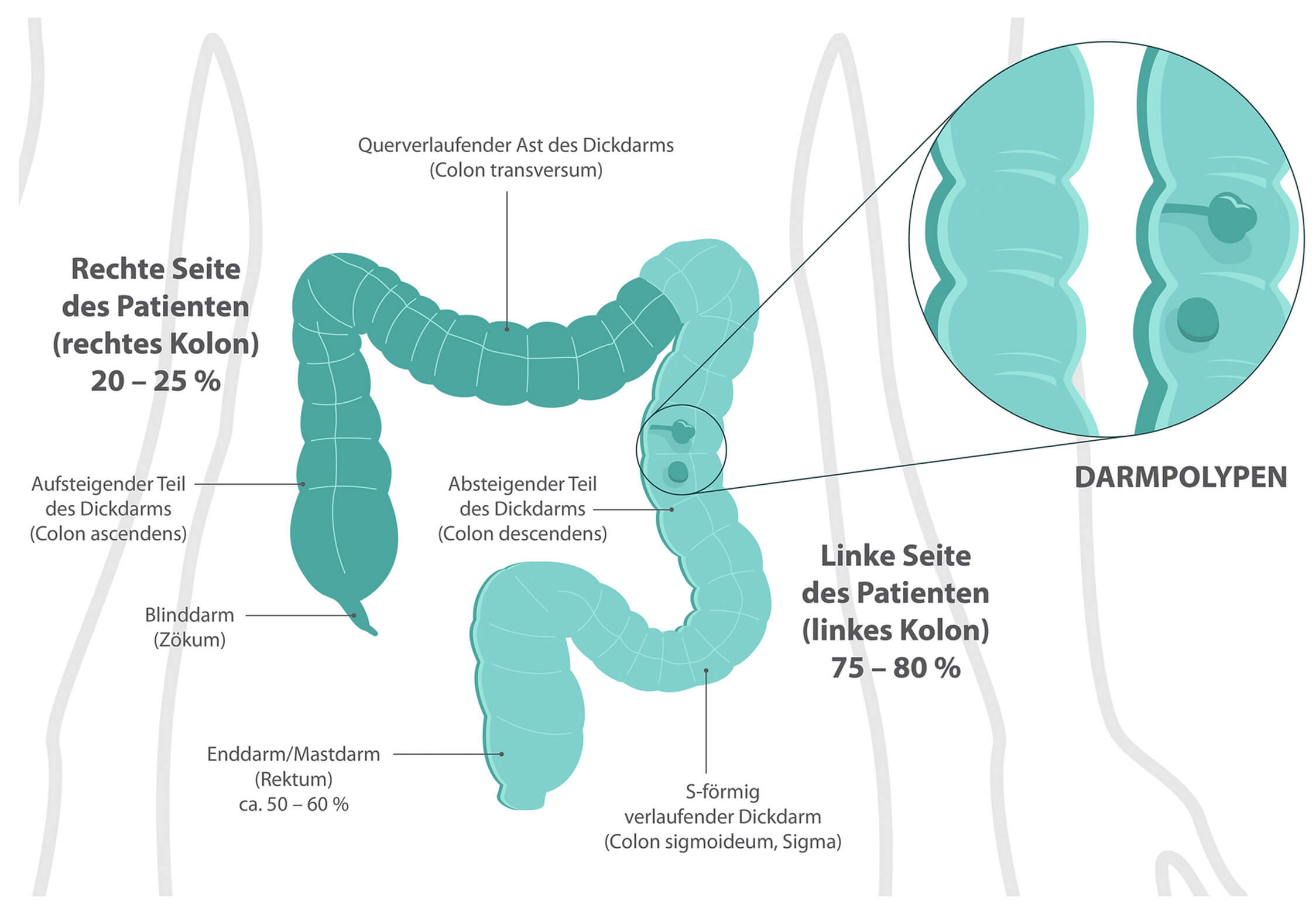
Die meisten kolorektalen Karzinome entstehen sporadisch und entwickeln sich aus primär gutartigen Vorstufen.1,2 Sie entstehen deutlich häufiger auf der linken Seite des Kolons und im Rektum als auf der rechten Seite. Bei genetischen Syndromen kann sich dieses Verhältnis jedoch umkehren.7
Etwa 20 bis 30 % der Tumoren treten familiär gehäuft auf, ohne dass eine konkrete genetische Ursache bekannt ist.8 Nur selten erkranken Personen an monogen verursachten kolorektalen Tumoren. Diese machen weniger als 5 % der CRCs aus. Zu ihnen zählen beispielsweise das hereditäre nicht-polypöse Kolonkarzinom-Syndrom (HNPCC, Lynch Syndrom) sowie familiäre Polyposis-Syndrome.8
Verschiedene Faktoren erhöhen die Wahrscheinlichkeit, im Laufe des Lebens an einem malignen Darmtumor zu erkranken. In der Normalbevölkerung ohne genetische oder familiäre Belastung liegt dieses Risiko bei etwa 6 %.9 Zu den Risikofaktoren zählen neben dem HNPCC sowie den familiären Polyposis-Syndromen:9
Kolorektale Karzinome verursachen meist erst Symptome, wenn sie bereits fortgeschritten sind. Die Beschwerden sind hierbei abhängig von der Größe und Lokalisation des Tumors und lassen sich wie folgt unterscheiden:4
Bei Verdacht auf ein kolorektales Karzinom werden zur Bestätigung der Tumorerkrankung und zum Staging eine Reihe von Untersuchungen empfohlen. Zu diesen zählen beispielsweise eine digitale rektale Untersuchung, eine komplette Koloskopie, eine CEA-Bestimmung sowie eine Sonographie des Abdomens und ein Röntgen-Thorax.3,4,8 Darüber hinaus können, abhängig von der Lokalisation des Tumors, weitere Untersuchungen indiziert sein, zum Beispiel eine starre Rektoskopie, eine MRT (CT) des Beckens, eine Endosonographie des Rektums sowie eine CT des Thorax.3,4,8 Zudem spielt die Bestimmung von Biomarkern und Genexpressionsprofilen bei der Diagnostik kolorektaler Karzinome eine entscheidende Rolle. Wichtige Gene, die vor der Therapiewahl untersucht werden sollten, sind u.a. KRAS, NRAS und BRAF. Zusätzlich sollte überprüft werden, ob der Tumor eine Stabilität oder Instabilität bestimmter kurzer, sich wiederholender DNA-Abschnitte aufweist – die sogenannte Mikrosatelliteninstabilität (MSI).4 Hierdurch können die Tumoren immer genauer differenziert und die Therapie individuell geplant werden.
Zur Therapie des kolorektalen Karzinoms stehen verschiedene Therapieoptionen zur Verfügung. Diese umfassen neben der Operation und Chemotherapie auch zielgerichtete Therapien und die Therapie mittels Checkpoint-Inhibitoren, einem immunonkologischen Wirkprinzip. Die bestmögliche Behandlung wird stadien-, biomarker- und risikoabhängig ausgewählt und erfolgt in Deutschland nach der S3-Leitlinie „Kolorektales Karzinom“8 und den Onkopedia-Leitlinien „Kolonkarzinom“4 und „Rektumkarzinom“3. Bis vor Kurzem wurde die Behandlung von Darmkrebs im Stadium IV als rein palliativ betrachtet. Heute ist bekannt, dass bei etwa 25 % der Patient:innen ein kurativer Therapieanspruch besteht, insbesondere wenn die Metastasen operativ entfernt werden können.4
Referenzen:
Bildquelle:iStock/MicroStockHub
ONC-DE-2500007