Ein Forscherteam hat eine neuartige Strategie zur Behandlung von Chorea Huntington entwickelt, bei der die Proteine im betroffenen Genabschnitt weiterhin funktionstüchtig bleiben.
Chorea Huntington ist eine erbliche neurodegenerative Erkrankung, die durch eine Mutation im Huntington-Gen verursacht wird: Dabei wird dem Protein ein erweiterter Abschnitt der Glutamin-Aminosäuren – auch Polyglutamin genannt – hinzugefügt. Die Spaltung in der Nähe des gestreckten Polyglutamins im Huntingtin-Gen ist ursächlich für die Erkrankung. Da das Huntington-Protein jedoch für die Entwicklung und die Funktion des Gehirns erforderlich ist, ist es schwierig lediglich die krankheitsverursachenden Proteine zu beseitigen und gleichzeitig die Funktionen zu erhalten.
Einem Forscherteam ist dies jedoch nun gelungen: Die Ergebnisse ihrer Studie zeigten, dass es eine Methode gibt, um das mutierte Huntington-Protein in eine krankheitsfreie Form umzuwandeln und gleichzeitig seine ursprüngliche Funktion zu erhalten.
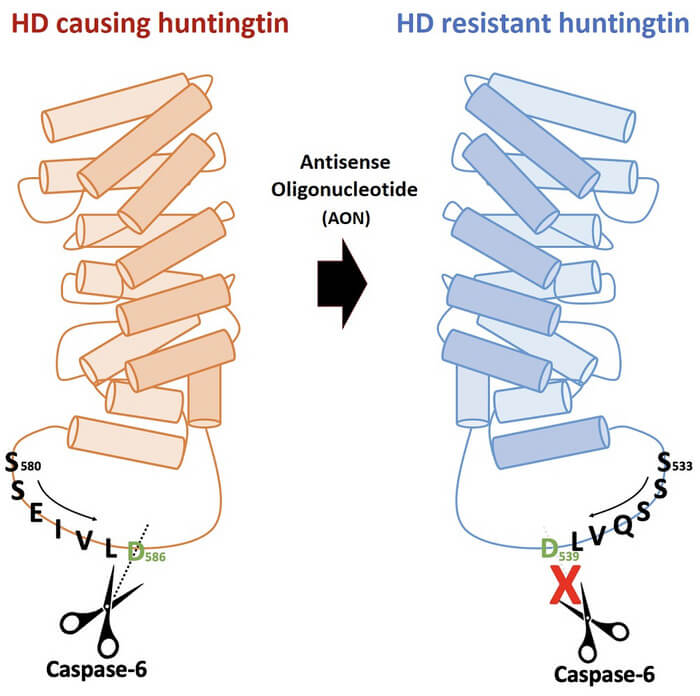
Das durch Antisense-Oligonukleotid (AON) induzierte Huntingtin-Protein, das gegen die Caspase-6-Spaltung resistent ist, verursacht keine Huntington-Krankheit und behält gleichzeitig die normalen Funktionen bei. Quelle: Hyeoungjun Kim, et al., 2022.
Die Wissenschaftler fanden heraus, dass die Huntington-Delta-12-Form, die mit Hilfe von Antisense-Oligonukleotiden hergestellt wurde, gegen die Spaltung durch Caspase-6 resistent ist. Diese ist für die Entstehung der Abspaltungen an den Enden des Proteins verantwortlich und somit für die Hauptursache der Erkrankung. Die Strategie könnte nun zur Entwicklung neuer Therapiemöglichkeiten beitragen, da sie Krankheitssymptome lindert und gleichzeitig die Funktionen des normalen Huntingtons aufrechterhält.
Dieser Text basiert auf einer Pressemitteilung des The Korea Advanced Institute of Science and Technology (KAIST). Hier findet ihr die Originalpublikation. Bildquelle: Katie McNabb, unsplash.