Die Cystinose gehört zu den seltenen lysosomalen Speicherkrankheiten und ist gekennzeichnet durch eine Anhäufung von Cystin in den Lysosomen. Dies verursacht Schäden in verschiedenen Organen und Geweben – zunächst hauptsächlich in den Nieren und den Augen. Sie ist genetisch bedingt und tritt auf, wenn die ursächliche Mutation über beide Elternteile, also autosomal-rezessiv, vererbt wird. Geschätzt leiden weltweit zwischen einer Person von 100.000 und einer Person von 200.000 an einer Cystinose.3
Grundsätzlich wird zwischen drei verschiedenen Formen von Cystinose unterschieden, wobei 95 % der Betroffenen an der infantilen Form, der nephropathischen Cystinose leiden.3 Die anderen beiden Formen sind die juvenile und die adulte (okuläre, nicht-nephropathische) Form.
Auslöser für eine Cystinose sind Mutationen im CTNS-Gen 17p, meist in der Chromosomenregion 17p13. Das CTNS-Gen kodiert das lysosomale Membranprotein Cystinosin, sodass die daraus resultierenden Mutationen dazu führen, dass Cystin nicht mehr in normalem Umfang aus den Zellen abtransportiert wird. Je stärker sich die zugrundeliegende Mutation auf die Funktionsfähigkeit des Cystinosins auswirkt, desto schwerer ist die Erkrankung. Mutationen, die dem Protein eine Restfunktion erlauben, resultieren in milderen Krankheitsverläufen, also der juvenilen oder okulären Form.4
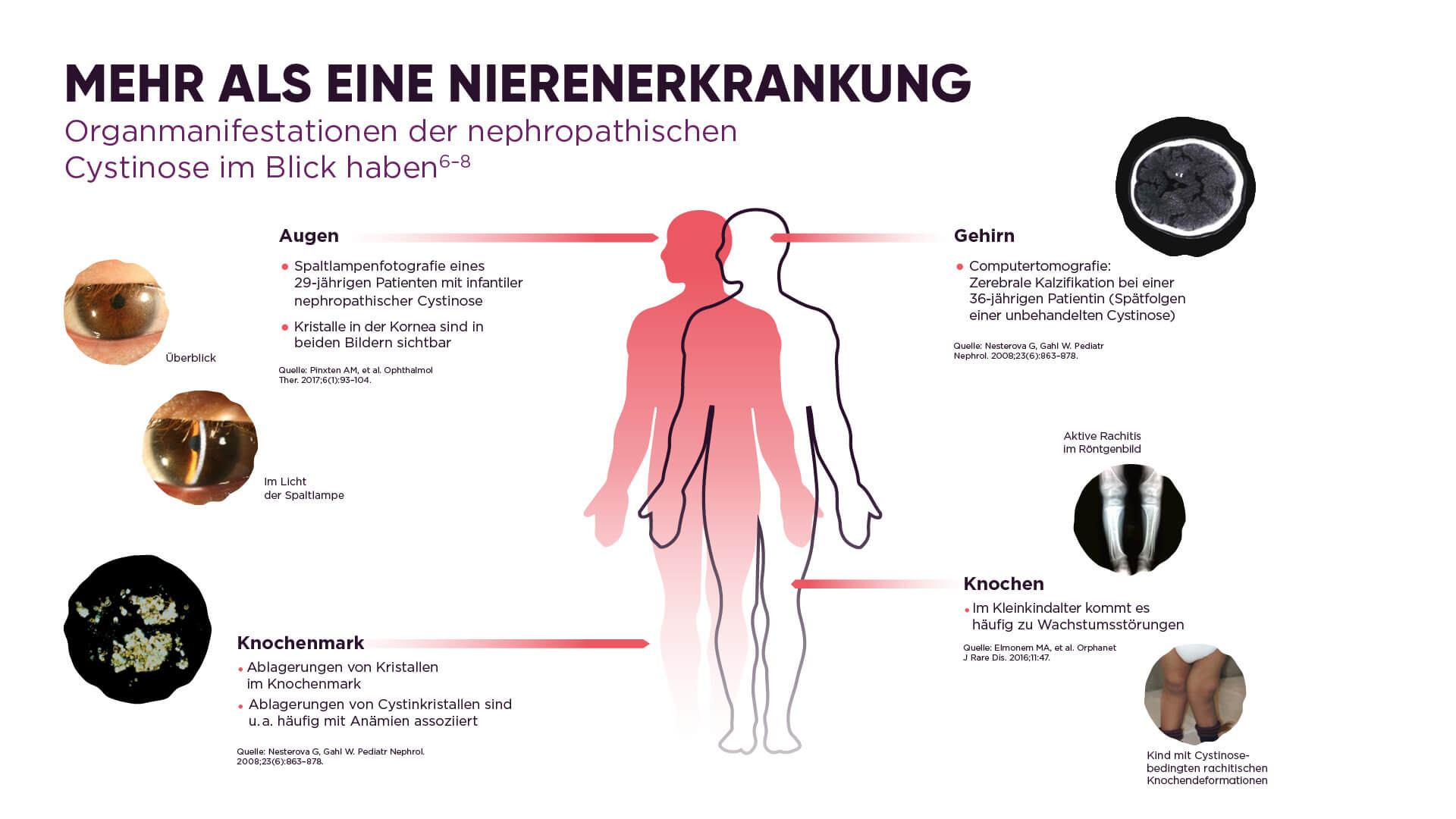
Bei der nephropathischen Cystinose sind zunächst besonders die Nieren betroffen. Hier kommt es zu schweren Funktionsstörungen. Bereits im Kleinkindalter tritt dabei häufig das Fanconi-Syndrom auf.3 Auffällig ist dabei, dass die Betroffenen sehr häufig Wasser lassen müssen und dementsprechend sehr durstig sind. Weil sie vermehrt Wasser und Elektrolyte ausscheiden, wird neben der körperlichen Entwicklung auch ihr Wachstum gestört. Typisch sind auch Augenbeschwerden, wie zum Beispiel Lichtempfindlichkeit. Im weiteren Verlauf einer nephropathischen Cystinose können Symptome wie Muskelschwäche oder Schilddrüsenunterfunktion auftreten und weitere Organgruppen können betroffen sein (s. Abb. 1).5
Abb. 1: Organmanifestationen der nephropathischen Cystinose
Bei Verdacht auf eine Cystinose gibt es drei wichtige Diagnosemethoden, die meistens miteinander kombiniert werden:
Nach der Diagnose gilt es, früh eine Therapie einzuleiten. Denn auch wenn mit heutigen Mitteln keine Heilung möglich ist, hat sich durch verschiedene Behandlungsmöglichkeiten die Prognose für Patient*innen in den letzten Jahrzehnten glücklicherweise deutlich verbessert. Organschäden durch Cystin-Ablagerungen können mittels einer Cystin-entspeichernden Therapie verzögert werden. Diese Behandlungen ermöglichen den Abbau von Cystin in den Lysosomen und den Abtransport der Reaktionsprodukte.11 Ein möglichst früher Therapiebeginn ist entscheidend für den Krankheitsverlauf. Ohne Therapie verstarben die meisten Patient*innen bereits vor dem 30. Lebensjahr.12
Egal, um welche Erkrankung es sich handelt: Die ausführliche Versorgung mit wichtigen Informationen ist unerlässlich. Deswegen haben wir unsere Rare Diseases Webseite ins Leben gerufen. Hier finden Sie nicht nur Informationen zur ganzheitlichen Cystinosetherapie, Symptomen, Diagnoseverfahren und Möglichkeiten des Therapiemanagements, sondern auch hilfreiche Materialien für Ihre Patient*innen sowie Angehörige:
(Er)kennen Sie Cystinose!
Bildnachweis: iStock.com/Colorfuel Studio