Mit einer Prävalenz zwischen 1:40.000 und 1:117.000 weltweit zählt Morbus Fabry (MF) zu den seltenen lysosomalen Speicherkrankheiten. Doch eventuell ist MF gar nicht so selten, denn manche Screeningprogramme zeigen sogar eine Inzidenz von bis zu 1:1250.
MF wird X-chromosomal vererbt. In Folge sind besonders Männer betroffen, bei denen das durchschnittliche Manifestationsalter zwischen 3 und 10 Jahren liegt. Auch Frauen können an MF erkranken, zeigen jedoch häufig einen milderen Verlauf und entwickeln im Durchschnitt Symptome im Alter zwischen 6 und 15 Jahren.
Die Ursache der Krankheit ist eine Mutation im GLA-Gen, das für das lysosomale Enzym alpha-Galactosidase A (AGLA) kodiert. Unter physiologischen Bedingungen ist AGLA an dem Abbau von sogenannten Glykosphingolipiden (GLS) beteiligt. GLS sind wichtige Bestandteile der Zellmembran.
Insgesamt sind mehr als 1000 Varianten dieser Mutation bekannt, die eine verminderte oder fehlende Enzymaktivität verursacht. Resultierend aus diesen Defekten können bestimmte GLS, darunter v.a. Gb3 und lyso-Gb3, nicht oder nur unzureichend in den Lysosomen abgebaut werden und reichern sich in diesen Organellen an.
Unbehandelt ist die Lebenserwartung der Betroffenen aufgrund von Niereninsuffizienz, Kardiomyopathie oder Schlaganfällen im Vergleich zur gesunden Bevölkerung stark reduziert, bei Männern bis zu 20 Jahre und bei Frauen bis zu 10 Jahre.
Morbus Fabry ist eine multisystemische Erkrankung, die eine Vielzahl von Symptomen verursacht.
Es wird angenommen, dass die Ablagerungen von Gb3 und lyso-Gb3 Entzündungen, Fibrosen und ischämische Hypertrophien verursachen. In Folge entstehen permanente Gewebsschäden.
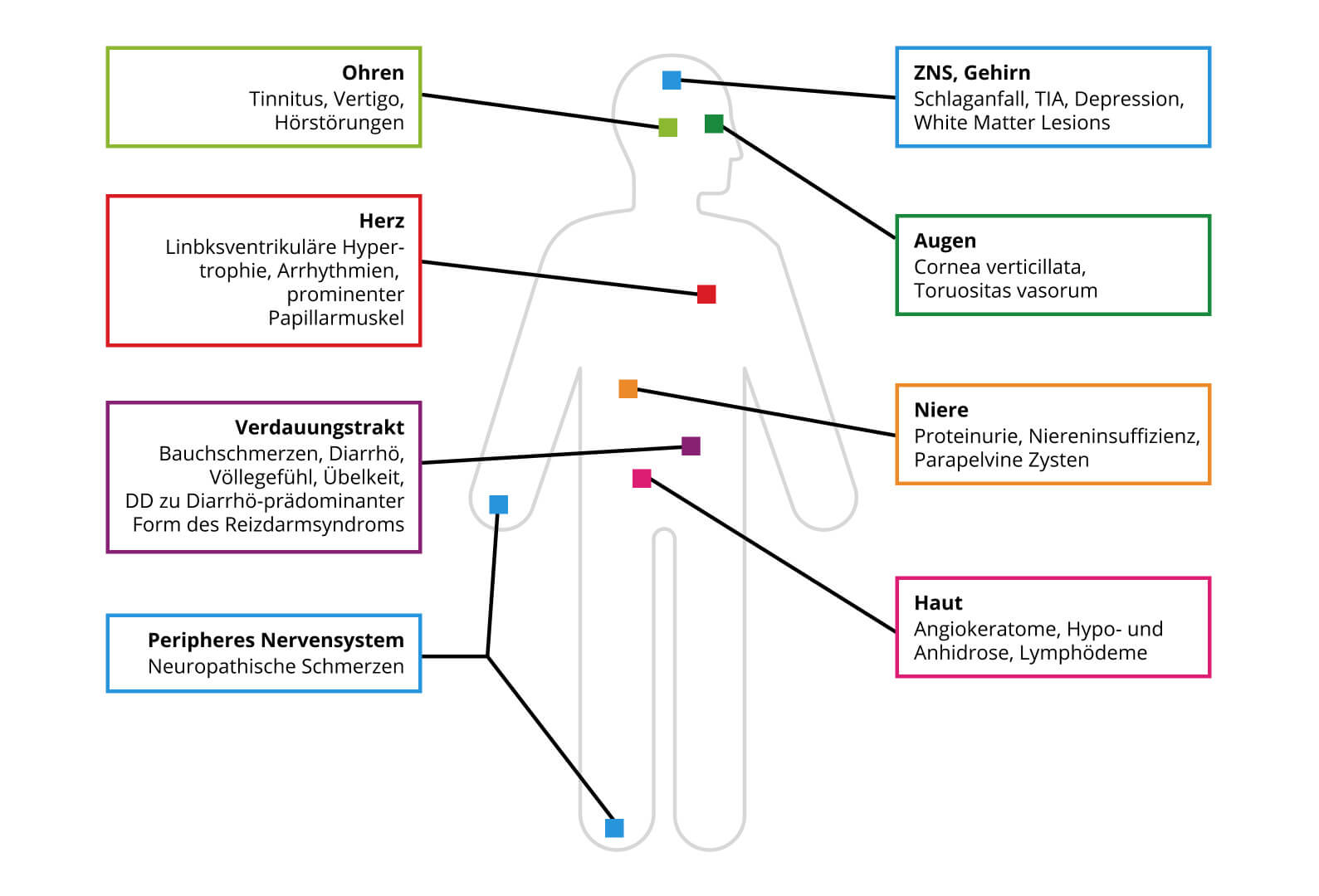
Diese Ablagerungen finden sich besonders in den folgenden Organen und Strukturen:
Die genaue Verteilung der Ablagerungen ist abhängig von der vorliegenden Genvariante und dem Geschlecht.
Der akute Verlauf von Morbus Fabry ist höchst variabel. Besonders häufig treten Angiokeratome sowie eine Cornea verticillata auf. In der Regel treten mit zunehmendem Alter zusätzliche Symptome auf und die Schwere der bestehenden Symptome nimmt zu (Abb.1).
Abb. 1: Typische betroffene Organe und Symptome bei Morbus Fabry.
Haupttodesursachen bei Patient:innen mit M. Fabry sind Niereninsuffizienz, plötzlicher Herztod durch Herzrhythmusstörungen und Schlaganfall.
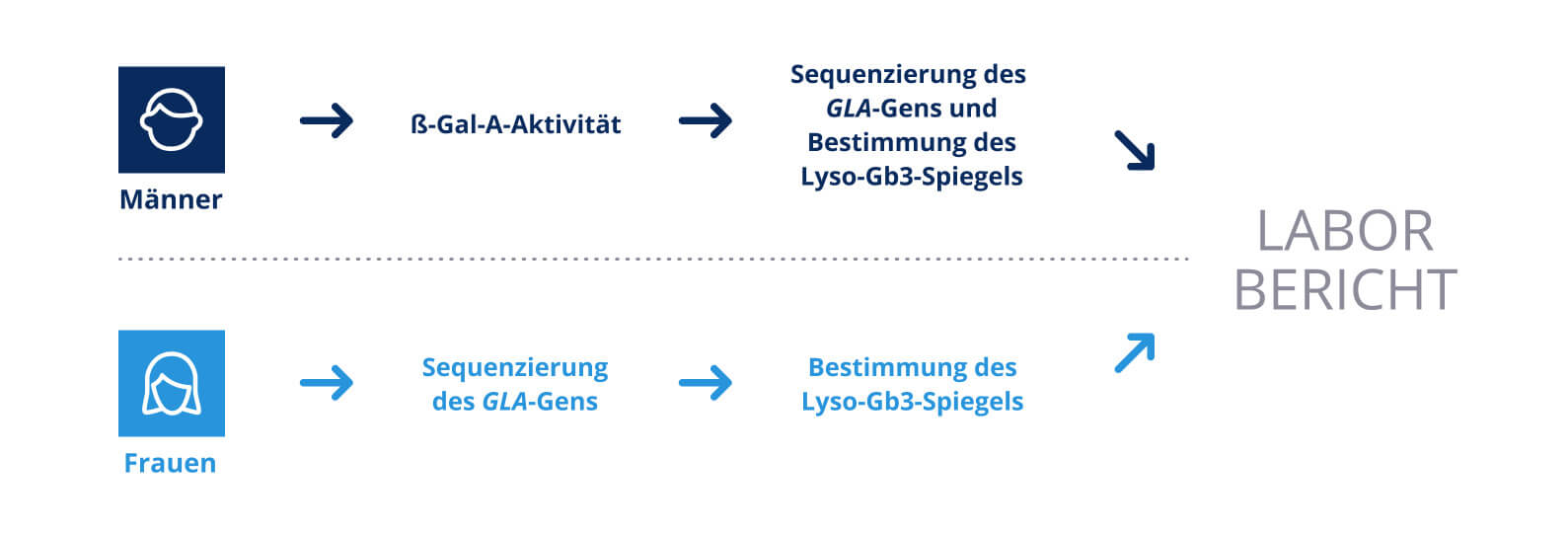
Die Diagnose von Morbus Fabry ist aufgrund der unspezifischen und vielfältigen Symptome oft eine Herausforderung. Ein Verdacht auf die Erkrankung sollte bei einer Kombination der oben genannten Symptome geweckt und in einer gründlichen Differenzialdiagnose abgeklärt werden. Dabei gestaltet sich die Diagnostik geschlechtsabhängig:
Bei Männern kann bei Verdacht auf MF die AGLA-Enzymaktivität in Leukozyten mithilfe eines Trockenbluttests bestimmt werden. Bei einer AGLA-Aktivität zwischen 0 und 24 % des unteren Referenzwerts wird dabei von MF ausgegangen. Dabei korreliert eine Enzymaktivität im höheren Bereich mit einem milderen Verlauf der Krankheit. Um die Diagnose final zu bestätigen, ist eine molekulargenetische Analyse des GLA-Gens und eine Bestimmung des Lyso-Gb3-Spiegels in einem akkreditierten Labor nötig. Zudem erlaubt diese Methode auch die eindeutige Identifikation der vorliegenden Genvariante (Abb.2).
Bei Frauen kann es aufgrund der zufälligen Inaktivierung des X-Chromosoms vorkommen, dass hämatopoetische Zellen noch eine Wildtyp-Kopie des GLA-Gens tragen. In Kombination mit weiteren genetischen und nicht-genetischen Faktoren wird davon ausgegangen, dass dies einer der Gründe dafür ist, dass 20 bis 30 % der erkrankten Frauen im Blut dennoch eine AGLA-Enzymaktivität im Normalbereich aufweisen. Daher ist bei Frauen nur eine molekulargenetische Untersuchung gefolgt von einer Bestimmung des Lyso-Gb3-Spiegels zur Diagnosestellung geeignet (Abb.2).
Abb. 2: Diagnostik von Morbus Fabry abhängig vom Geschlecht.
Bei Verdacht auf MF können Tests unter anderem bei spezialisierten Diagnostikinitiativen angefordert werden.
Anschließend an die Diagnose sollte die Überweisung an ein spezialisiertes Morbus Fabry-Kompetenzzentrum erfolgen. So werden eine umfassende Fabry-Diagnostik und ein frühzeitiger Therapiebeginn ermöglicht.
Nach der Bestätigung des MF ist eine Eingangsuntersuchung der typischerweise betroffenen Organe und Organsysteme nötig, um etwaige Organschäden zu erkennen. Dazu werden verschiedene bildgebende Verfahren benutzt, etwa die Kardio-Magnetresonanztomographie zur Untersuchung des Herzens, sowie die Sonographie und kraniale MRTs zur Untersuchung von PNS und ZNS. Im Weiteren wird das Fortschreiten der Krankheit in jährlichen Verlaufskontrollen überwacht.
Die genaue Therapieeinstellung sollte in Morbus Fabry-Kompetenzzentren stattfinden. In diesen Kompetenzzentren findet die Behandlung der Symptome durch unterschiedliche Fachgruppen koordiniert statt. Je nach vorliegender Mutation stehen unterschiedliche Therapien zur Verfügung, wie beispielsweise eine Enzymersatztherapie (EET) oder eine Chaperontherapie.
Neben der Behandlung des Enzymmangels sind oftmals auch weitere Medikation und Therapien nötig, um Begleiterkrankungen zu adressieren. Infolgedessen werden Patient:innen von einem interdisziplinären Team betreut.
Wollen Sie noch mehr zum Thema Morbus Fabry erfahren? Dann finden Sie weitere Informationen hier.
EXA/DE/FAB/0276_09/2024