Wissenschaftlern ist es nun im Zuge einer Studie gelungen, die Ursachen einer seltenen erblichen Nierenerkrankung – einer familiären Form des renalen Fanconi-Syndroms – aufzuklären.
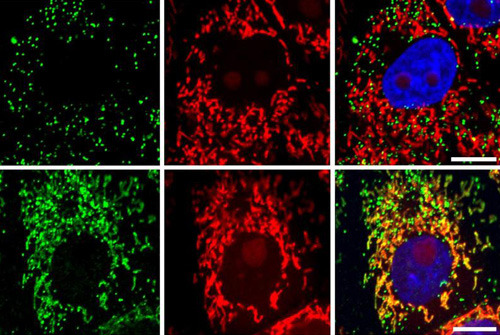
Die Nieren erfüllen ihre lebenswichtige Ausscheidungsfunktion, indem sie für die Urinproduktion zunächst Flüssigkeit aus dem Blutplasma abfiltrieren. Im Anschluss werden die Menge und die Zusammensetzung des Filtrats in einem komplexen System von Kanälchen verändert. Abbauprodukte, die der Körper ausscheiden muss, werden im Urin angereichert. Glucose, Aminosäuren, Phosphat und andere Stoffe, die der Körper noch benötigt, werden allerdings erneut unter Energieverbrauch aus dem Urin aufgenommen bzw. rückresorbiert. Beim sogenannten renalen Fanconi-Syndrom ist dieser Rücktransport beeinträchtigt und die Patienten verlieren die wichtigen Nährstoffe mit der Ausscheidung des Urins. Die Folge ist eine Rachitis-ähnliche Symptomatik. Ausgelöst werden kann das Krankheitsbild durch verschiedene Nieren-schädigende Substanzen; beispielsweise durch Arzneimittel, die bei der HIV-Therapie eingesetzt werden, oder durch eine Reihe von vererbbaren Gendefekten. Ein Forscherteam um Dr. Markus Reichold, Dr. Jörg Reinders und Prof. Dr. Richard Warth (alle Universität Regensburg) hat nun gemeinsam mit einer englischen Arbeitsgruppe um den Kinderarzt und Genetiker Prof. Dr. Robert Kleta (University College London) herausgefunden, dass bei der erblichen Form des renalen Fanconi-Syndroms ein völlig neuartiger Krankheitsmechanismus vorliegt. So weisen die betroffenen Personen eine Mutation des Proteins EHHADH auf, das in bestimmten Zellorganellen (Perixosomen) vorkommt und dort am Abbau von Fettsäuren beteiligt ist. Überraschenderweise machten die Forscher die Entdeckung, dass ein vollständiges Fehlen von EHHADH zu keiner Nierenfunktionsstörung führt. Eine Erkrankung mit der vererbbaren Form des renalen Fanconi-Syndroms kann demnach nicht durch einen einfachen Funktionsverlust von EHHADH bedingt sein. Warth und seine Kollegen machten sich vor diesem Hintergrund auf die Suche nach den tieferliegenden Ursachen der Erkrankung. Oben das normale EHHADH-Protein (grün), ohne lokale Überlappung mit Mitochondrien (rot). Unten mutiert: die Lokalisation überlappt mit Mitochondrien (Mischfarbe aus rot und grün, Bild rechts unten). © Bild: Universität Regensburg Ihre Untersuchungen führten zu einem verblüffenden Ergebnis: Damit die Nierenzellen korrekt funktionieren können, müssen die verschiedenen Bausteine der Zelle nicht nur vorhanden und intakt, sondern auch an der richtigen Stelle platziert sein. Eine Mutation von EHHADH führt in diesem Zusammenhang zu einer falschen „Adressierung“. Das mutierte Protein gelangt durch den Adressierungsfehler in die Mitochondrien. Dorthin fehlgeleitet, bedingt EHHADH – wie ein falsches Zahnrad in einem Getriebe – eine Störung der Energieproduktion. Den Nierenzellen fehlt es in diesem Fall an der nötigen Energie, um wichtige Nährstoffe aus dem Urin zurücktransportieren zu können. Formen des renalen Fanconi-Syndroms, die durch Gendefekte verursacht werden, sind nach heutigem Wissensstand noch nicht dauerhaft heilbar. Die neuen Erkenntnisse zu den Ursachen der Erkrankung können aber dazu beitragen, neuartige und personalisierte Therapien zu entwickeln, die auch den spezifischen genetischen Ursachen der einzelnen Patienten gerecht werden. Originalpublikation: Mistargeting of Peroxisomal EHHADH and Inherited Renal Fanconi's Syndrome Markus Reichold et al.; N Engl J Med, DOI: 10.1056/NEJMoa1307581; 2014