Das Dup15q-Syndrom, kurz für Chromosom 15q11.2-q13.1 Duplikationssyndrom, ist eine seltene genetische Erkrankung, die unter anderem durch Entwicklungsverzögerungen, Muskelhypotonien und Autismus-Spektrum-Störungen gekennzeichnet ist. Ursächlich sind Duplikationen in der sogenannten Prader-Willi/Angelman-kritischen Region (PWACR).
Die betroffene Region auf Chromosom 15 wird als Prader-Willi/Angelman-kritische Region (PWACR) bezeichnet, da Deletionen des Abschnitts das Prader-Willi-Syndrom und Angelman-Syndrom auslösen. Zu den Genen, die in diesem Abschnitt kodiert sind, zählen u.a. die Flippase ATP10A, die Ubiquitinligase UBE3A sowie verschiedene GABA-A-Rezeptor-Untereinheiten. Die Duplikation einzelner Gene an dem Genlokus führt nicht zum Dup15q-Syndrom.
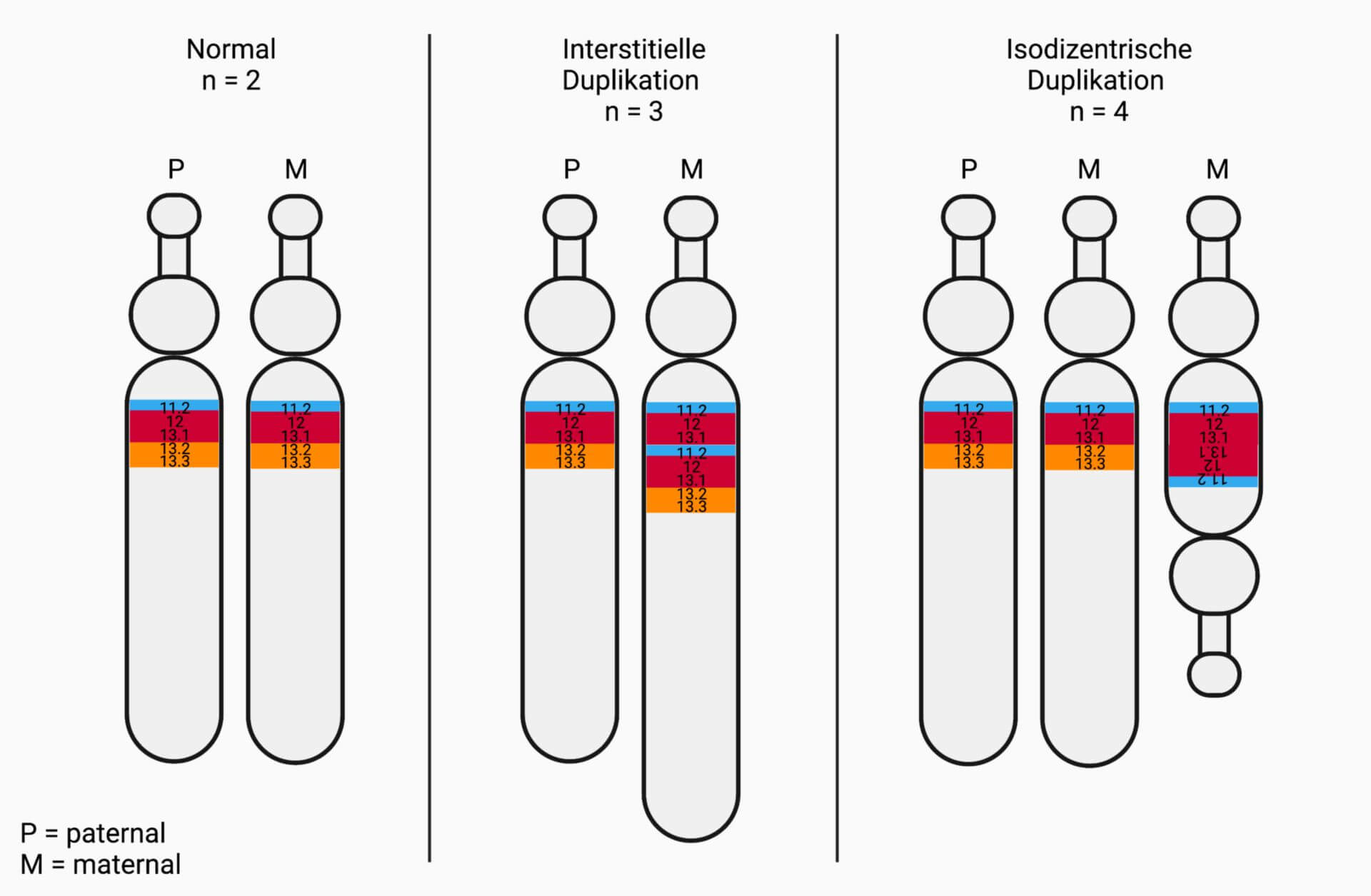
Die Chromosomenaberration, die beim Dup15q-Syndrom am häufigsten auftritt (~ 80 % d.F.), ist ein isodizentrisches Chromosom 15, kurz idic(15). Dieses liegt als zusätzliches Chromosom neben den beiden normalen Kopien vor und enthält zwei gespiegelte PWACRs, die sich zwischen zwei Centromeren befinden. Individuen haben dementsprechend vier Kopien des Segments.
Bei ~ 20 % der Betroffenen befindet sich die Duplikation innerhalb des langen Arms (q-Arm) auf einem der beiden Chromosomen. Diese Form wird als interstitielle Duplikation, kurz int dup(15), bezeichnet. Hierbei handelt es sich meist um eine Neumutation. Im Vergleich zu idic(15) liegen nur drei Kopien der PWACR vor, entsprechend weisen die Patienten meist mildere Verläufe auf.
Quelle: Lusk et al. Maternal 15q Duplication Syndrome, GeneReviews®
Autor: Janica Nolte, DocCheck, erstellt mit BioRender.com; lizenziert unter CC BY-NC-SA 3.0