Das Hutchinson-Gilford-Syndrom ist eine autosomal-dominante Erkrankung verschiedener Gewebe, die zu einem massiven und sehr früh einsetzenden Alterungsprozess (Progerie) von Haut, Skelett und Blutgefäßen in frühster Kindheit führt. Von manchen Autoren wird auch ein autosomal-rezessiver Erbgang angegeben.
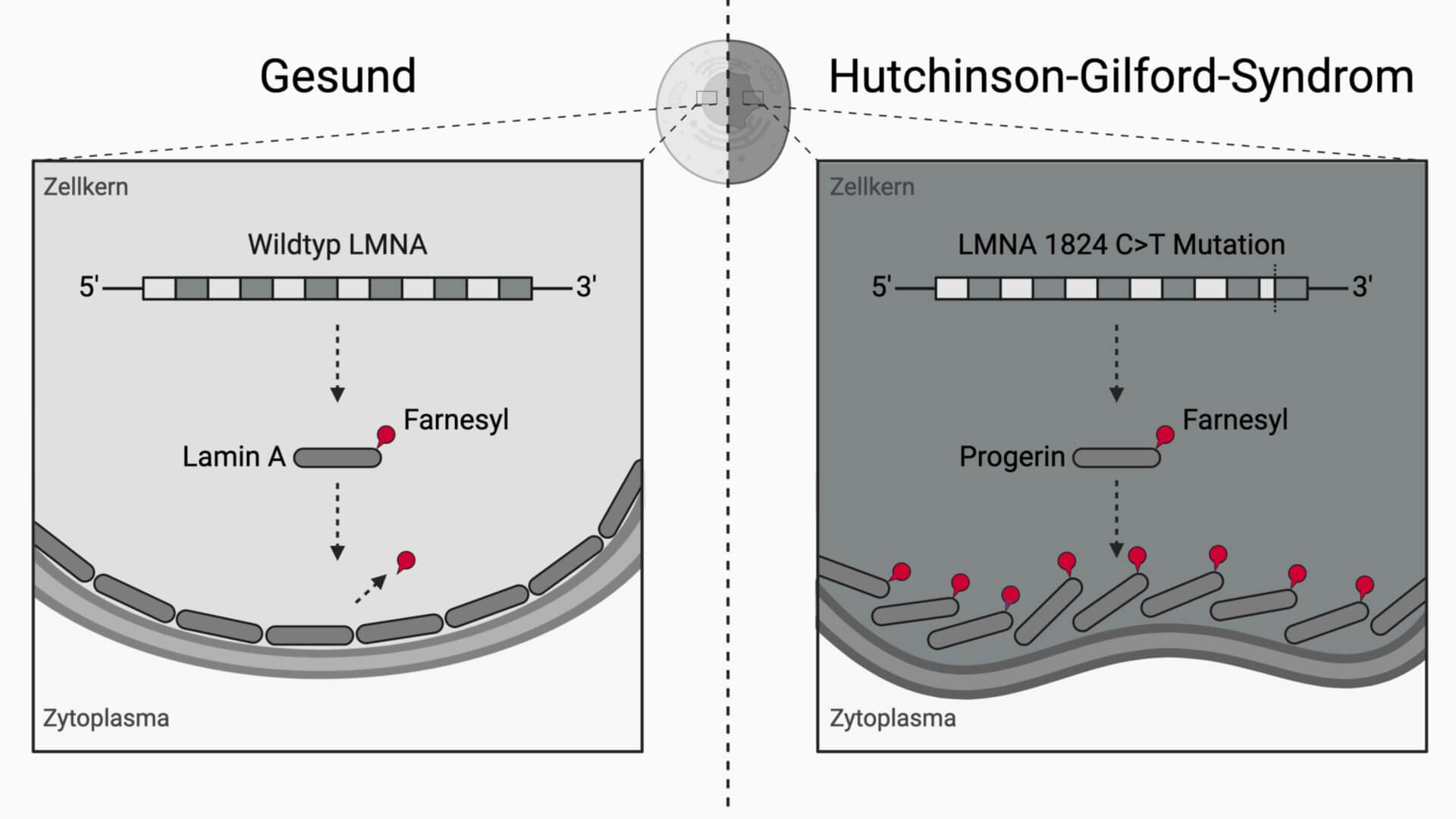
Dem autosomal-dominant vererblichen Hutchinson-Gilford-Syndrom liegt ein Defekt des LMNA-Gens auf einem Allel von Chromosom 1 (1q21.3) vor, das für das Protein Lamin A/C kodiert. Lamin A/C ist ein in der Kernmembran der meisten Zellen vorkommendes Protein, das als Anknüpfungspunkt des Filamentsystems und vermutlich auch als Regulator der Gentranskription fungiert.
Bei einem überwiegenden Teil der Patienten mit Hutchinson-Gilford-Syndrom ist das Codon 608 verändert, sodass eine kryptische Splicestelle aktiviert wird. Dabei gehen 150 Nukleotide des Exons 11 verloren, was zum Verlust von 50 internen Aminosäuren im späteren Protein, das Progerin genannt wird, führt. Progerin gelangt zwar in den Nucleus und lagert sich dort an die Kernhülle an, das Lamingerüst kann allerdings nicht korrekt gebildet werden. Dies resultiert in einer Deformation des Nucleus, die sich durch eine lobuläre Form auszeichnet.
Autor: Janica Nolte, DocCheck; adaptiert von “Hutchinson-Gilford Progeria Syndrome LMNA Mutation Mechanism” by BioRender.com (2024); lizenziert unter CC BY-NC-SA 3.0